
ما هي الأخطاء الوراثية في الاستقلاب (IEM)؟
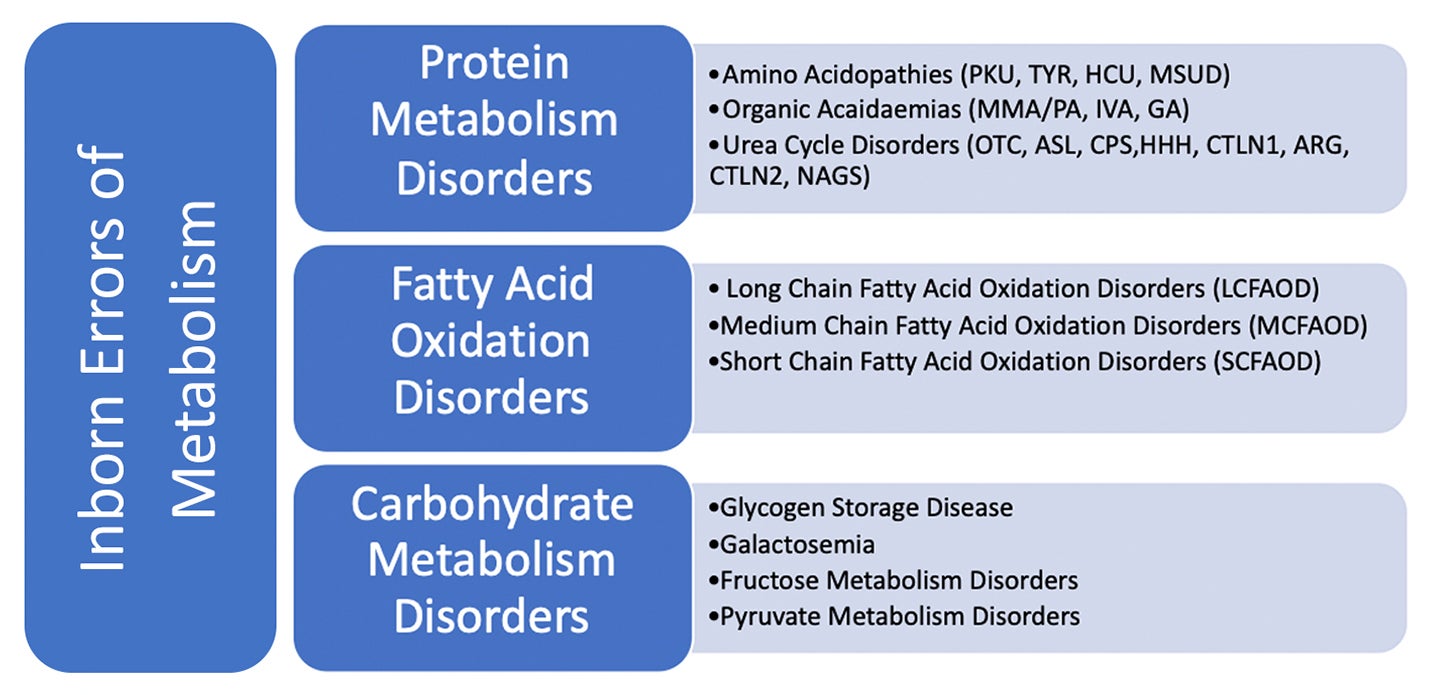
العيوب الوراثية في الاستقلاب (IEM) هي اضطرابات جينية موروثة حيث يؤدي عيب إنزيم معين إلى تعطيل التمثيل الغذائي للبروتين أو الدهون أو الكربوهيدرات. وبسبب انخفاض نشاط الإنزيم أو غيابه في هذه الاضطرابات ، تتراكم مركبات معينة إلى مستويات سامة داخل الجسم. يؤدي أيضاً إلى عدم قدرة الجسم على إنتاج مركبات معينة بشكل كافٍ، مما يؤدي إلى حدوث نقص، ويمكن أن تؤدي هذه الاضطرابات الأيضية، إذا تركت دون علاج، إلى مجموعة من النتائج الطبية والنمائية، من ضعف الإدراك وفشل الأعضاء وحتى الموت.
يمكن تخفيف العديد من العواقب السلبية للاستقلاب IEM عن طريق الاكتشاف المبكر والعلاج الذي يمكن أن يشمل كلاً من الأدوية والتدخل الغذائي ، باستخدام الأطعمة للأغراض الطبية الخاصة المعروفة باسم FSMP's.
تعتبر العيوب الوراثية في الاستقلاب IEM نادرة على الصعيد الفردي ولكنها أكثر شيوعًا على الصعيد الجماعي (٣.0 -0.9٪ من سكان العالم (1)). يتم فحص العديد من العيوب الوراثية في الاستقلاب IEM عند الولادة من خلال اختبار وخز الكعب، ويسمح فحص حديثي الولادة بالتعرف المبكر وبدء العلاج الإداري الذي يحسن نتائج المرضى. أدت فعالية فحص حديثي الولادة إلى زيادة معدلات الإصابة بـ IEM، وبينما يوصى بالفحص، لكنه لا يتوفر في جميع البلدان.
فينيلكيتونوريا (PKU) هي واحدة من أشهر العيوب الوراثية (IEM) التي تتطلب إدارة غذائية ولها معدل حدوث عالمي يقارب ١ من كل ١٠,٠٠٠ ٳلي ١ من كل ١٢,٠٠٠
(2).
بيلة الفينيل كيتون هو اضطراب وراثي يتطلب وراثة جين بيلة الفينيل كيتون من كلا الوالدين وليس مرتبطًا بالجنس، وهذا ما يُعرف باسم وراثي متنحي. PKU هو اضطراب في استقلاب البروتين، ويرتبط بشكل خاص بالحمض الأميني فينيلالانين (الأحماض الأمينية هي اللبنات الأساسية للبروتين). يتميز PKU بنقص الإنزيم الكبدي المعروف باسم فينيلالانين هايدروكسيلاز الذي يحول فينيلالانين (Phe) إلى تيروسين (Tyr)، مما يؤدي إلى تراكم Phe ونقص Tyr في الدم.
عندما لا يتم علاج هذا يمكن أن يؤدي إلى مستويات سامة من Phe مما يؤدي إلى تلف الدماغ الدائم مما يسبب صعوبات التعلم والتغيرات السلوكية والرعشة والنوبات المرضية. يتسبب انخفاض مستويات Tyr في ضعف إنتاج الناقل العصبي ونقص الميلانين، مما يؤدي إلى بشرة شاحبة مميزة. يجب مراقبة مستويات Phe و Tyr في حالة فينيلكيتونوريا (PKU) عن طريق أخذ بقع دم منتظمة وإدارة تناول البروتين الطبيعي.
نظرًا لأن Phe هو حمض أميني أساسي، فهو ضروري لوظيفة الجسم الطبيعية ولا يمكن أن ينتجه الجسم. لتجنب تراكم Phe إلى مستويات سامة، لا يزال من الضروري استهلاك كمية صغيرة من البروتين الطبيعي المقاسة بإحكام لضمان تلبية متطلبات Phe ولكن لا يتم تجاوزها. الأفراد الذين يعانون من فينيلكيتونوريا (PKU) لديهم تفاوتات مختلفة في Phe اعتماداً على شدة حالتهم ومرحلة حياتهم ومعدل نموهم.
البروتين ضروري للنمو والإصلاح. الكميات الصغيرة من البروتين الطبيعي التي يستهلكها مرضى فينيلكيتونوريا (PKU) لا تكفي لتلبية متطلبات البروتين الإجمالية. يتم وصف المنتجات التي تسمى بدائل البروتين (PS) من أجل تحقيق مستويات البروتين المطلوبة للمرضى.
تستخدم مصطلحات مختلفة لوصف بدائل البروتين بما في ذلك الغذاء الطبي والصيغة والمكملات الغذائية. تحتوي بدائل البروتين على ما يعرف بـ" مكافئ البروتين "مما يعني أن المنتج يحتوي على جميع الأحماض الأمينية الموجودة داخل البروتين ولكن مع الأحماض الأمينية المخالفة، في هذا حالة Phe تمت إزالتها كلياً أو تم تقليلها بشكل كبير. تتم إضافة مغذيات إضافية إلى PS بسبب الطبيعة التقييدية للنظام الغذائي من أجل منع نقص التغذية PS. الأطعمة المصنعة منخفضة البروتين متاحة أيضًا للاستخدام في الإدارة الغذائية لفينيلكيتونوريا (PKU)، بالإضافة إلى اضطرابات ااستقلاب الأخرى. وتشمل هذه الأطعمة الأساسية مثل الخبز منخفض البروتين، والمعكرونة وبدائل الحليب التي تتيح الشعور بالحالة الطبيعية للنظام الغذائي بينما أيضًا توفير طاقة إضافية لدعم التحكم العام في الاستقلاب.
يمكن أن يؤدي التشخيص المبكر والتدخل الغذائي إلى تحسن كبير في نتائج المرضى (3).
يمكن العثور على مزيد من المعلومات حول IEM وإدارة النظام الغذائي في Vitaflo | Nestle Health Sience (nestlehealthscience.xx <- يمكن أن يحل محله موقع شركتك المحلي إذا كان لديك ركن Vitaflo).
يجب على أي شخص مصاب بالـ IEM يرغب في الحصول على مزيد من المعلومات حول الاضطراب والإدارة والنتائج ، طلب المشورة الطبية من الطبيب أو أخصائي التغذية.
مراجع
- Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. European Journal of Human Genetics 2019 28:2 [Internet]. 2019 Sep 16 [cited 2021 Oct 21];28(2):165–73. Available from: https://www.nature.com/articles/s41431-019-0508-0
- Dixon M, MacDonald A, White F, Stafford J. Disorders of Amino Acid Metabolism, Organic Acidaemias and Urea Cycle Disorders. Clinical Paediatric Dietetics: Fourth Edition [Internet]. 2014 Nov 17 [cited 2021 Oct 13];381–525. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/9781118915349.ch17
- Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Archives of Disease in Childhood [Internet]. 2007 Mar 1 [cited 2021 Oct 15];92(3):213–8. Available from: https://adc.bmj.com/content/92/3/213
اقرأ المزيد عن فيتافلو