
What are inborn errors of metabolism (IEM)?
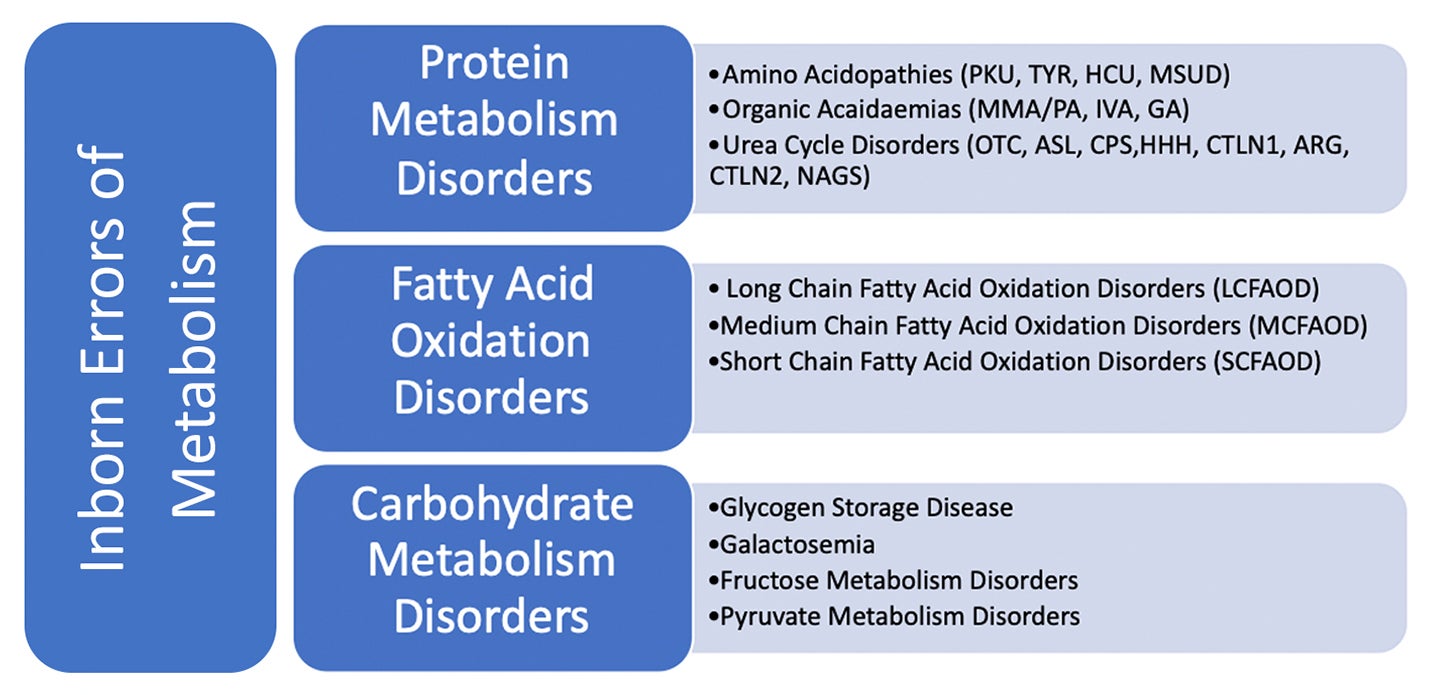
Inborn errors of metabolism (IEM) are inherited genetic disorders whereby a specific enzyme defect disrupts normal protein, fat, or carbohydrate metabolism. Due to decreased or absent enzyme activity in these disorders, specific compounds accumulate to toxic levels within the body. This can also result in the body being unable to adequately produce specific compounds that it requires, leading to deficiencies. These metabolic disturbances, if left untreated, can result in a range of medical and developmental outcomes, from cognitive impairment, organ failure and even death.
Many of the negative consequences of IEM can be mitigated by early detection and treatment which can include both drug and dietary intervention, utilising foods for special medical purposes known as FSMP’s.
Individually IEM’s are rare but are collectively more common (3.5 -5.9% of the worldwide population (1)). Many IEM’s are screened for at birth via the Heel Prick Test, newborn screening allows early identification and initiation of management treatment improving patient outcomes. The effectiveness of newborn screening has resulted in an increase in incidence rates of IEM’s and whilst screening is recommended, not all countries have this in place.
Phenylketonuria (PKU) is one of the most common IEM that requires nutritional management and has a global incidence rate of approximately 1 in 10,00 to 1 in 12,000 (2).
PKU is an inherited disorder that requires the PKU gene to be inherited from both parents and is not sex linked, this is known as autosomal recessive. PKU is a protein metabolism disorder, specifically related to the amino acid Phenylalanine (Amino acids are the building blocks of protein). PKU is characterised by a deficiency of hepatic enzyme known as phenylalanine hydroxylase that converts phenylalanine (Phe) to tyrosine (Tyr), resulting in a build-up of Phe and a deficiency of Tyr in the blood.
When untreated this can result in toxic levels of Phe leading to irreversible brain damage causing learning difficulties, behavioural changes, tremors and seizures. Low Tyr levels cause impaired neurotransmitter production and melanin deficiency, resulting in characteristic pale complexion. Both Phe and Tyr levels need to be monitored in PKU by taking regular blood spots and managing natural protein intake.
As Phe is an essential amino acid, it is required for normal bodily function and cannot be produced by the body. To avoid Phe building up to toxic levels, a small tightly measured amount of natural protein must still be consumed to ensure Phe requirements are met but not exceeded. Individuals with PKU have different Phe tolerances depending on the severity of their condition, life stage and growth rate.
Protein is essential for growth and repair. The small amounts of natural protein consumed by PKU patients is not enough to meet total protein requirements. Products called protein substitutes (PS) are prescribed in order to achieve a patients required protein levels.
Various terms are used to describe protein substitutes including medical food, formula and supplement. Protein substitutes contain what is known as ‘protein equivalent’ meaning that the product contains all of the amino acids found within protein but with the offending amino acid, in this case Phe, removed entirely or greatly reduced. Additional nutrients are added to PS due to the restrictive nature of the diet in order to prevent nutritional deficiency. There are two main sources of protein substitute available for PKU - amino acid passed PS and glycomacropeptide (GMP) based PS. Manufactured low protein foods are also available for use in dietary management of PKU, as well as other metabolic disorders. These include staple foods such as low protein bread, pasta and milk alternatives allowing a sense of normality to the diet whilst also providing additional energy to support overall metabolic control.
Early diagnosis and dietary intervention can significantly improve patient outcomes (3).
More information on IEM and dietary management can be found at Vitaflo | Nestlé Health Science (nestlehealthscience.com)
Anyone affected by IEM wanting more information about the disorder, management, and outcomes, should seek medical advice from their clinician / dietitian.
References:
- Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. European Journal of Human Genetics 2019 28:2 [Internet]. 2019 Sep 16 [cited 2021 Oct 21];28(2):165–73. Available from: https://www.nature.com/articles/s41431-019-0508-0
- Dixon M, MacDonald A, White F, Stafford J. Disorders of Amino Acid Metabolism, Organic Acidaemias and Urea Cycle Disorders. Clinical Paediatric Dietetics: Fourth Edition [Internet]. 2014 Nov 17 [cited 2021 Oct 13];381–525. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/9781118915349.ch17
- Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Archives of Disease in Childhood [Internet]. 2007 Mar 1 [cited 2021 Oct 15];92(3):213–8. Available from: https://adc.bmj.com/content/92/3/213
Read more about vitaflo
Related products
